The P2D2 (Predicted Powder Diffraction Database) is built up by calculating the powder patterns corresponding to the predicted crystal structures from the latest PCOD update [1]. The job is done partly by ZEFSA II (898.707 entries) and for the GRINSP data (163.520 entries) by the CIF2POW software [2] able to read a large multiple CIF and to provide the corresponding powder patterns in a format described below.
Example of a powder pattern in the P2D2 :
15 PCOD1000014 Click here to see the CIF Al F3 10.2102 10.2102 7.2408 90.000 90.000 90.000 P 4/N M M 6 99 5.9063 409. 1 0 1 5.1125 283. 1 1 1 5.1051 197. 2 0 0 4.1723 999. 2 0 1 3.8623 835. 1 2 1 3.6204 35. 0 0 2 3.6099 238. 2 2 0 3.4122 453. 0 1 2 3.2363 336. 1 1 2 3.2306 570. 2 2 1 3.2287 544. 3 1 0 3.0801 530. 0 3 1 2.9532 102. 2 0 2 etc, etc 1.2596 54. 0 4 5 1.2586 70. 5 3 4 1.2501 12. 1 4 5 1.2475 82. 8 1 1 1.2408 58. 3 3 5 1.742 Ref: A. Le Bail, J. Appl. Cryst. 38 (2005) 389-395.
Format of the data in the P2D2 text file :
line 1 : No of the phase in the P2D2 (from 1 to 107449)
line 2 : No of the phase in PCOD
this is a 7 digits number after the four letters PCOD
ex : PCOD1000000
That number allows for retrieving the atomic coordinates in PCOD
at http://www.crystallography.net/pcod/
line 3 : chemical formula
ex : Si O2 or B4 Si3 O12 , Si3 Ti O9,
etc
There is a space between each group of (element + number)
line 4 : cell parameters a, b, c, alpha, beta, gamma
line 5 : probable space group (it is a prediction...)
ex : P -4 M 2 or P 4/N M M, I 41/M,
etc
- there is a space between the symmetry elements
- sometimes noted "unknown" (data coming from the
first GRINSP version)
line 6 ; a symmetry code number (1-14)
1 = Cubic
2 = Body-centered cubic
3 = Face-centered cubic
4 = Rhombohedral (if not hexagonal setting)
5 = Hexagonal
6 = Tetragonal
7 = Body-centered tetragonal
8 = Orthorhombic
9 = Body-centered orthorhombic
10 = Face-centered orthorhombic
11 = Base-centered orthorhombic
12 = Monoclinic
13 = Base-centered monoclinic
14 = Anorthic
line 7 : number N of pairs [d(A) and Intensity] + hkl
lines 8 and following : N lines of pairs d(A) and I + the hkl
- Imax = 999.
- intensities at delta(d) < 0.0002 A were added
- calculated by L x F**2 x multiplicity
(L = Lorentz factor)
- for X-ray, wavelength = 1.54056 A
- peaks with I < 10 are discarded
(all models are optimized in the P1 space group,
so that there could be weak-intensity peaks
violating the existence conditions of the "probable"
space group...)
- the d, I, hkl list stops either at 99 lines or at dmax = 1A
next line : I/Icor
last line : reference
The P2D2 contains >1.000.000 powder diffraction patterns calculated from predicted open framework crystal structures of inorganic oxydes and fluorides. Silicates, phosphates and sulfates of Ti, V, Ga, Nb, Zr; zeolites, etc. These data are produced by two distinct crystal structure prediction computer programs :
ZEFSA II (Michael W. Deem and David J. Earl) : 898.707 SiO2 zeolites entries selected according to energy criteria (GULP calculations) among a largest dataset of ~2.7M entries.
GRINSP (Armel Le Bail) [3-4]: 163.520 entries, lists of formulations are available corresponding to the 2009 PCOD update, together with classifications according to the framework density (...-FD.txt files), the quality R-factor (...-R.txt files) the complete lists of connectivity sequences (CS) (...-con.txt files) and the multiple CIF corresponding to each series. GRINSP produces N- or N-N'-connected 3D frameworks. The values explored for N-N' are 3, 4, 5, 6, corresponding to polyhedra such as triangles, tetrahedra, pentahedra and octahedra connected exclusively by corners, forming either binary or ternary compounds [5-7].
The main purpose of the P2D2 is to allow for identification of unknown compounds which would even not be recognized by a search-match [8] through the powder diffraction file (ICDD - PDF) (containing essentially the powder data for known compounds).
The P2D2 cannot be used directly with any search-match software, up to know. There is the need to compile the P2D2 in order to prepare the proprietary files as required by the various search-match computer programs (EVA-Bruker, Jade, Match from Crystal Impact, Highscore from PANalytical, etc).
Up to now, the P2D2 was compiled to be used with the EVA search-match software (Bruker) and Highscore (PANalytical).
A demonstration of the performances with EVA is available inside of a .doc file for MS Word 2000. See also the powerpoint file corresponding to a recent conference in Brazil. A paper was published [9] in the Powder Diffraction Journal, supplementary issue June 2008. The main conclusion is that the search-match can be successful only if the predicted cells parameters are different by no more than 1% from the those of the real compound. One should note that a cell parameters difference of less than 1% will not always be fulfilled by the current structure prediction computer programs, so that, it is recommended, in cases you know the cell parameters of your unidentified compound, to check the whole PCOD database by using larger ranges of cell parameters (+- 5% for instance) or cell volumes. Of course, if you don't know your cell parameters (unindexed powder pattern), then only the search-match option through the predicted powder patterns will have some chances of success - if the predictions are accurate enough...
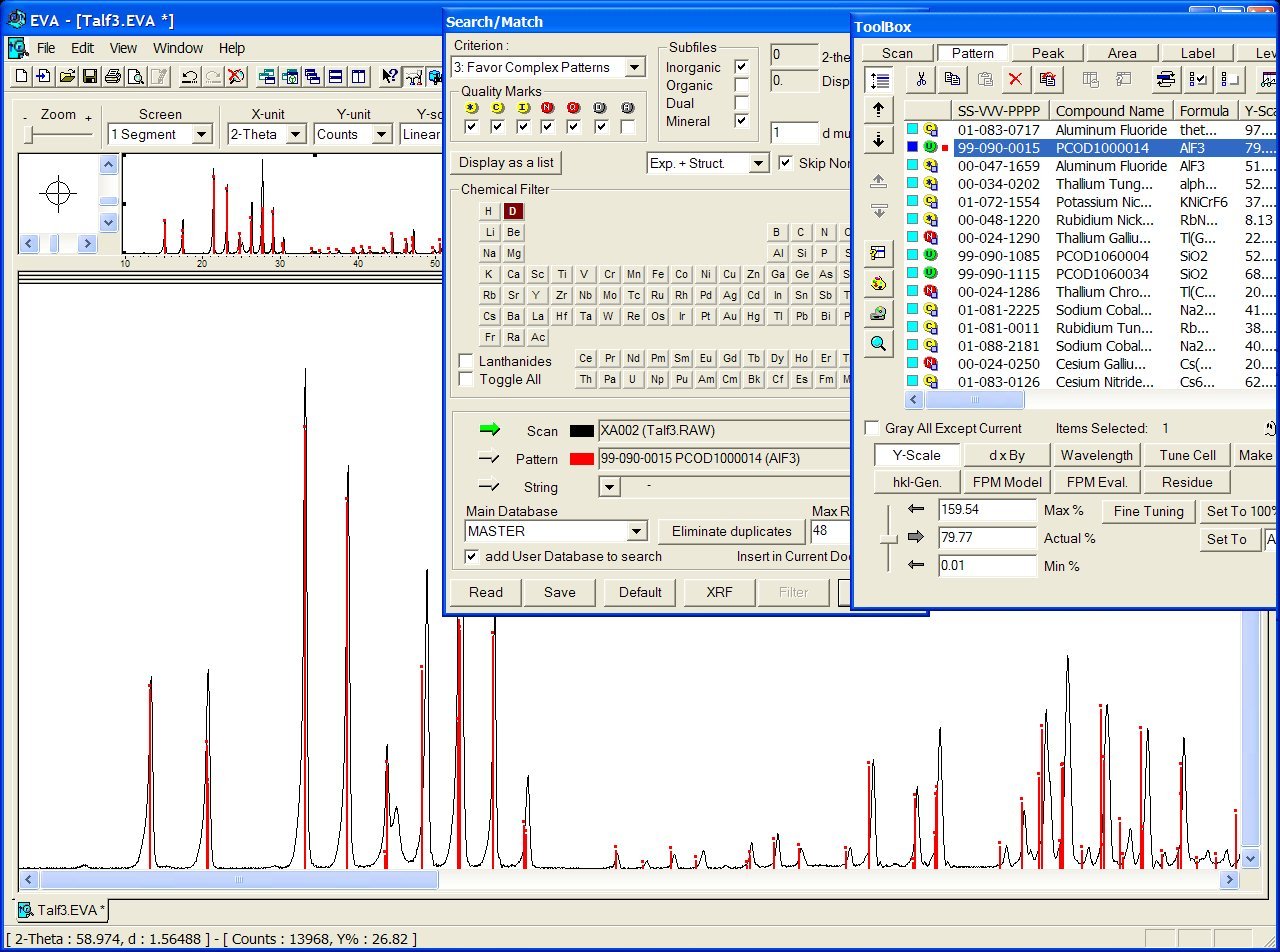
In case of a successful identification, you will obtain the CIF filenumber in PCOD (see the figure below sith the file PCOD1000014). Then go to the PCOD database in order to obtain the CIF (use 1000014 or PCOD1000014 in the text search box).
Note also that realizing the search-match in your powder pattern 2-theta range < 40 or 30° will increase your chances of success with the P2D2, because the peak position at low diffraction angle is less sensitive to large cell parameter discrepancies (this is clear on the figure below). The knowledge of your zeropoint is also essential.
PCOD
The >1.000.000 entries in the PCOD are delivered in full open access
under the CIF format.
A search engine allows for retrieval of the PCOD entry number, formula,
cell parameters, volume, space group, text :
http://www.crystallography.net/pcod/
or (frequently updated first) :
http://sdpd.univ-lemans.fr/cod/pcod/
The CIFs from ZEFSA-II have names between 8000001.cif and 8313568.cif
for SLC-refined models and 9000001 to 9585139 for the BKS-refined models.
The CIFs from GRINSP have N******.cif names with N different from 8.
P2D2
The P2D2 (~1.000.000 powder patterns) may also be delivered as a text file for compilation purposes for other search-match software on demand. If your usual search-match program does not give access to the P2D2, ask to your search-match software provider.
The files necessary for EVA are also available here. The previous P2D2 version (~100.000 entries) could be compiled as a "user database" through the PDFMAINT satellite program. However, difficulties were encountered with the P2D2 update to ~1.000.000 entries. It was only found possible to cut the whole database in several parts containing close to 100.000 entries each :
Zeolites SLC data : part 1, part
2, part 3, part
4 (tetrahedra only);
Zeolites BKS data : part 1, part
2, part 3, part
4, part 5, part
6 (tetrahedra only);
GRINSP data : part 1 (zeolites and frameworks
with one sort of polyhedra, ~60.000 entries), part
2 (structure candidates combining two kinds of polyhedra, titanosilicates,
etc, ~100.000 entries).
This limitation does not occur with data from ICSD included in EVA because the compilation process is different (Master Database - not User Database).
So, unfortunately, to check your experimental data, you need to repeat 11 times the job by the selection of these 11 "user databases", one after the other. See how this is realized with EVA.
Contact : A. Le Bail
If you are interested in the addition of particular families of compounds, isostructural with other families already inside of the PCOD, the structures may be built on demand by the GRINSP software - if possible (N- or N-N' connected 3D nets).
More development of the GRINSP software is intended in order to allow for other connectivities than by corner-sharing (edge, face, mixed). Hole location, filling them with appropriate ions in order to ensure electrical neutrality. Using of the valence bond theory. Etc.
A question is to keep or not several descriptions of the same structure in different space groups and cells, as they may appear during the ZEFSA II / GRINSP predictions. In PCOD, the choice was made to keep only one model corresponding to a given connectivity sequence, selected for its highest symmetry and best R factor (so that each zeolite model is unique, etc).
[1] A. Le Bail. PCOD : The Predicted Crystallography Open Database.
http://www.crystallography.net/pcod/
[2] A. Le Bail. PPP - Powder Pattern Prediction, IUCr Commission on Powder Diffraction Newsletter 31 (2004) 51-53.
[3] A. Le Bail. Inorganic structure prediction with GRINSP, J. Appl.
Cryst. 38 (2005) 389-395. (PDF)
http://www.cristal.org/grinsp
[4] A. Le Bail. Geometrically restrained inorganic structure prediction : GRINSP, IUCr Computing Commission Newsletter 4 (2004) 37-45.
[5] A. Le Bail & F. Calvayrac. Hypothetical AlF3 crystal structures, J. Solid State Chem., 179 (2006) 3159-3166. (PDF)
[6] A. Le Bail. Inorganic structure prediction: too much and not enough, Solid State Phenomenon, 130 (2007) 1-6.
[7] A. Le Bail. Predicted corner-sharing titanium silicates, Z. Kristallogr., suppl. 26 (2007) 203-208.
[8] J-M. Le Meins, L.M.D. Cranswick and A. Le Bail. Results and conclusions of the internet based Search/Match Round Robin 2002, Powder Diffraction, 18 (2003) 106-113.
[9] A. Le Bail, Frontiers between crystal structure prediction and determination by powder diffractometry, Powder Diffraction, Suppl. 23(2) (2008) S5-S12. (PDF).